How to confirm your plasmid cloning worked for $1 per clone
A Plasmidsaurus hates this one weird trick, type post. As a follow up for my post on how to easily clone a plasmid, here is something I built out for my very own company (more soon) to streamline the cloning process. I hope that some people find this useful, and Plasmidsaurus doesn’t get angry at me. When I started in the lab 12 years ago (time flies) sequencing a plasmid costed about $80, took a ton of DNA, and multiple days. It is a small revolution that you can get the cost down to about $1 now.
8 months ago Plasmidsaurus flew me out to come to their SF site to talk about my work, the founder saw my cloning blog post. I guess it was kind of a job interview, which I was not informed of, but my plans were to go back to Boston after I finished my PhD. I was very impressed at their operation; they have robots doing some wild stuff and an extraordinarily efficient platform. I went and gave my spiel, I think they liked it as they booed when I said I’m going back to Boston. I remain blown away by them; and their service keeps getting better.
Anyway, when I clone a plasmid with the method described; I confirm the plasmid by doing an inverse PCR on the backbone in a colony PCR and sequencing the linear amplicon. This costs $15 per clone. Keeping my design-build cycles under a week is my highest priority, so I normally sequence 2-3 clones per plasmid depending on how badly I need the plasmid next day. Most of the time I’m cloning a few versions of the same thing, say 4 versions, meaning I need to sequence up to 12x clones, which runs me $180. In a well-funded lab that’s no big deal but starting a company on a $50k it adds up quickly.
Since I’m already running a PCR it’s natural to add barcodes at this stage. Demultiplexing is standard, and theoretically you don’t need all that many reads to determine the identity of your plasmid to single base pair confidence. About 5-7 reads should let you average over the whole plasmid and determine each base with confidence.
Plasmidsaurus has a service called “Premium PCR” where they return 3-5k reads of the DNA you sent for $30. That means you should be able to pool a bunch of these PCRs and pull them back apart to determine the identity of each well.
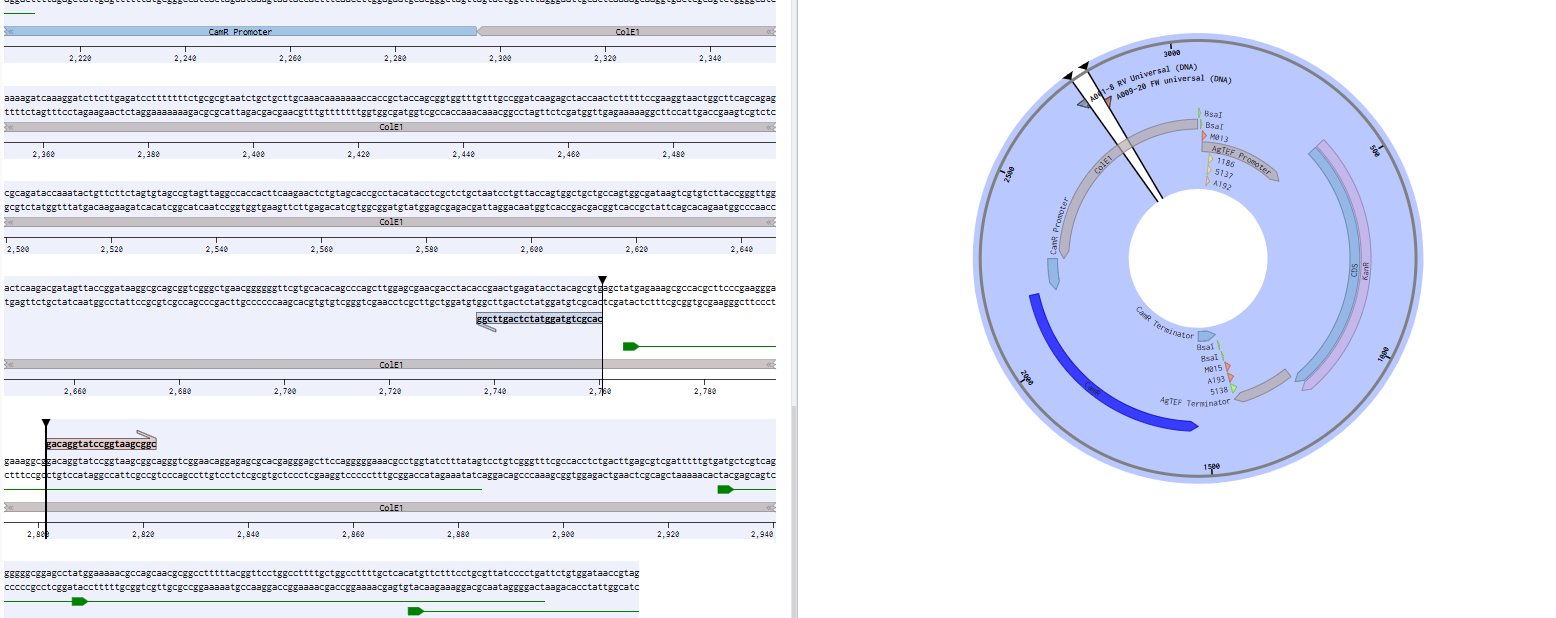
I started by gemini-vibe coding a test to look at the e.coli origin of replication of all of my plasmids and identify chunks that are common to every plasmid. 395/396 of them share the same high-copy origin, the other one is left out. I drop the code in here to illustrate, though it’s not really sharable because I’m terrible at coding. Anyone interested should be able to feed it into an LLM and adapt for their own purposes, though. It finds me some chunks of DNA which I use as primer binding sites, and will bind to all future plasmids I clone.
import os
import sys
import warnings
from Bio import SeqIO
from Bio.Seq import Seq
# ================= CONFIGURATION =================
# 1. Silence the circularity warnings
warnings.filterwarnings("ignore")
REF_ORIGIN_SEQ = "GGCCGCGTTGCTGGCGTTTTTCCATAGGCTCCGCCCCCCTGACGAGCATCACAAAAATCGACGCTCAAGTCAGAGGTGGCGAAACCCGACAGGACTATAAAGATACCAGGCGTTTCCCCCTGGAAGCTCCCTCGTGCGCTCTCCTGTTCCGACCCTGCCGCTTACCGGATACCTGTCCGCCTTTCTCCCTTCGGGAAGCGTGGCGCTTTCTCAATGCTCACGCTGTAGGTATCTCAGTTCGGTGTAGGTCGTTCGCTCCAAGCTGGGCTGTGTGCACGAACCCCCCGTTCAGCCCGACCGCTGCGCCTTATCCGGTAACTATCGTCTTGAGTCCAACCCGGTAAGACACGACTTATCGCCACTGGCAGCAGCCACTGGTAACAGGATTAGCAGAGCGAGGTATGTAGGCGGTGCTACAGAGTTCTTGAAGTGGTGGCCTAACTACGGCTACACTAGAAGGACAGTATTTGGTATCTGCGCTCTGCTGAAGCCAGTTACCTTCGGAAAAAGAGTTGGTAGCTCTTGATCCGGCAAACAAACCACCGCTGGTAGCGGTGGTTTTTTTGTTTGCAAGCAGCAGATTACGCGCAGAAAAAAAGGATCTCAAGAAGATCCTTTGATCTTTTCTACGGGGTCTGACGCTCAGTGGAACGAAAACTCACGTTAAGGGATTTTGGTCATGA"
WINDOWS_PATH = r"C:\Users\...\Plasmids"
# ==================== LOGIC ====================
def get_wsl_path(win_path):
path = win_path.replace("\\", "/")
if ":" in path:
drive, rest = path.split(":", 1)
if len(drive) == 1:
return f"/mnt/{drive.lower()}{rest}"
return path
def get_plasmid_sequence(filepath):
try:
if filepath.endswith(".gb") or filepath.endswith(".gbk"):
record = SeqIO.read(filepath, "genbank")
return record.seq
elif filepath.endswith(".fasta") or filepath.endswith(".dna"):
record = SeqIO.read(filepath, "fasta")
return record.seq
except Exception as e:
# Keep silent on read errors to avoid clutter, or print simple msg
pass
return None
def find_sequence_in_plasmid(plasmid_seq, query_str):
query_seq = Seq(query_str)
query_rev = query_seq.reverse_complement()
search_space = plasmid_seq + plasmid_seq
if query_seq in search_space: return True
if query_rev in search_space: return True
return False
def main():
folder_path = get_wsl_path(WINDOWS_PATH)
print(f"Scanning directory: {folder_path}")
if not os.path.exists(folder_path):
print("Path not found.")
return
# 1. Load Plasmids
plasmids = {}
try:
files = [f for f in os.listdir(folder_path) if f.endswith(".gb") or f.endswith(".gbk")]
except Exception:
print("Error listing files.")
return
print(f"Loading {len(files)} plasmids (warnings silenced)...")
for f in files:
full_path = os.path.join(folder_path, f)
seq = get_plasmid_sequence(full_path)
if seq: plasmids[f] = seq
if not plasmids:
print("No readable plasmids found.")
return
total_plasmids = len(plasmids)
# 2. Score Primers
print(f"\n--- Ranking 30bp Sites by Coverage (Total: {total_plasmids}) ---")
candidates = [] # Stores (score, start_index, sequence, list_of_missing_files)
window_size = 30
for i in range(len(REF_ORIGIN_SEQ) - window_size + 1):
candidate = REF_ORIGIN_SEQ[i : i + window_size]
missing_files = []
for name, p_seq in plasmids.items():
if not find_sequence_in_plasmid(p_seq, candidate):
missing_files.append(name)
score = total_plasmids - len(missing_files)
candidates.append((score, i, candidate, missing_files))
# 3. Sort by score (highest coverage first)
candidates.sort(key=lambda x: x[0], reverse=True)
# 4. Report Best Hits
# We will print the top hits, skipping overlaps for clarity
print(f"{'Cov':<5} | {'Pos':<5} | {'Sequence':<35} | {'Missing'}")
print("-" * 75)
last_printed_pos = -999
# Show top 10 distinct regions
shown = 0
best_candidate = candidates[0] if candidates else None
for score, start, seq_str, missing in candidates:
if score < (total_plasmids * 0.5):
break # Stop if less than 50% coverage
# Don't print every single base shift (e.g. pos 10, 11, 12).
# Only print if we are >15bp away from the last printed one.
if abs(start - last_printed_pos) > 15:
missing_str = str(len(missing))
if len(missing) > 0 and len(missing) < 4:
# If only a few are missing, list them roughly
missing_str = ",".join([m[:10]+"..." for m in missing])
print(f"{score}/{total_plasmids} | {start:<5} | {seq_str:<35} | {missing_str}")
last_printed_pos = start
shown += 1
if shown > 15: break
print("-" * 75)
# 5. Deep Dive on the Best Candidate
if best_candidate and len(best_candidate[3]) > 0:
print(f"\nBest candidate (Pos {best_candidate[1]}) failed in {len(best_candidate[3])} files:")
for m in best_candidate[3]:
print(f" - {m}")
print("\nTip: Check these files. They might have a point mutation or a different origin class.")
if __name__ == "__main__":
main()
Using this; I design 8x forward and 12x reverse primers, pointing outwards on the origin of replication. All bind the same complementary sites, but have different barcodes. The 8x forward primers will correspond to the row of a 96 well plate (A, B, C…H), and the 12x reverse primers correspond to the column of a 96 well plate (1, 2, 3…12). In the raw reads from plasmidsaurus, each read is now tagged on the ends with pieces of DNA that ID which well it came from. I ordered these 20 primers, and made a 96 well plate containing the unique combinations, diluted to 1.25 uM (each primer) in IDTE buffer. I keep this master plate at 4C. I’m a bit concerned at how long it will last as diluted primers decay, but we’ll see. It’s been good for a month so far.

Here’s an example FW primer with the barcode bolded: GTTGTGACGTATCACGCTGTAGGTATCTCAGTTCGG
And RV primer: AGTCAGCTACAGGACAGGTATCCGGTAAGCGGC
When I clone my plasmids, I pick colonies into 20 uL of water in a 96 well plate, and mix. This is used to inoculate either a 96 well deep well with LB + antibiotic, or 48 well deep well. I prefer 48 well, with 1.2 mL of media, so I can use 200 uL to make a glycerol stock the next day, and 1 mL to prep the plasmid. It is very important to keep the orientation the same between the PCR stage and the culture stage.
I prepare a master mix, scaled to the number of wells I am sequencing. I run 15 uL PCRs though it could be reduced further. In this setup each well costs about $0.60 to run.
PCR:
0.5 uL GXL poly
1.25 uL DNTPs
3 uL 5x MM
5.75 uL ddH2O
Add 10.5 uL per well of PCR strip or 96 well PCR plate
Then add:
2 uL colony
2.5 uL diluted primer mix
I run the PCR with standard GXL conditions. I do 2 min ext time, and 28x cycles. After the reaction I add 2 uL of loading dye to each well, and run 5 uL of the reaction on a gel. Then I pick wells which look promising by the correct size. These can be pooled, 5 uL per PCR, into one tube. Because the reaction added barcodes you will be able to pull back apart.
Below was a gel from some hairy cloning with lots of repeats and 11 kb plasmids.
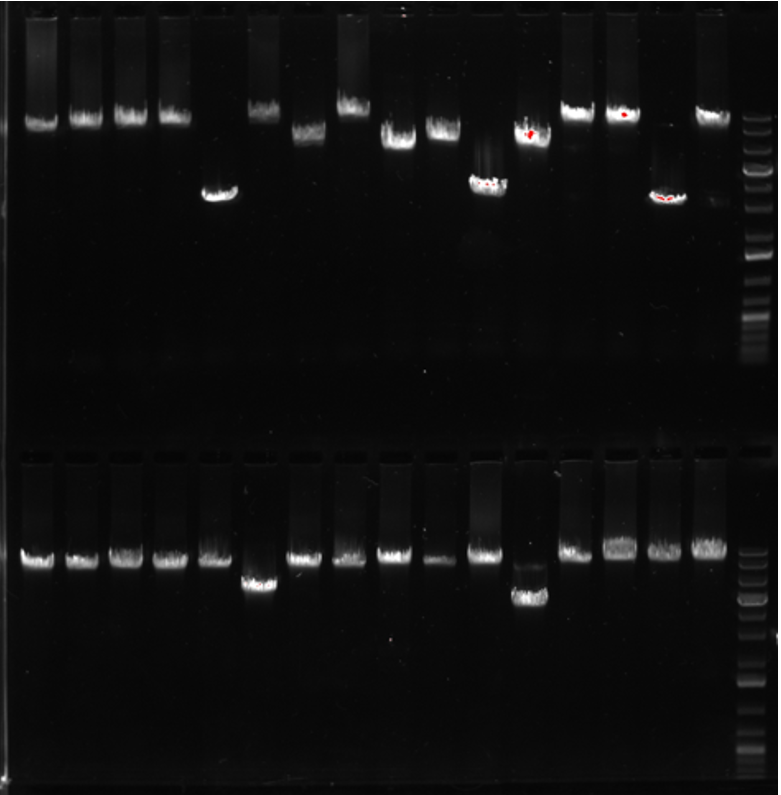
I send the pooled reactions to plasmidsaurus with the $5 cleanup add-on (I’m lazy). They have returned the results overnight 5/5 times I’ve run this, which is incredible. So far I get back ~5,000 reads. For some reason, 80% of these are junk, which comes out as <2 kb reads. Before doing demultiplexing/consensus construction I filter for reads above a certain length, depending on the plasmid I’m cloning. 5-6 kb is a reasonable cutoff point.
Then I had gemini write a script to demultiplex with cutadapt, take the longest read, and construct a consensus based on that using racon and minimap2. It outputs fasta files which contain the number of reads. A 0 error tolerance in the demultiplexing was critical for getting this to work and allowing for 1 error resulted in some reads being misallocated between wells. You lose reads but it’s ok. A lot of my python script is just dealing with windows subsystem for linux. Script below, same caveats as the primer binding site script.
import os
import subprocess
import sys
import shutil
from pathlib import Path
# ================= CONFIGURATION =================
INPUT_FILE_WIN = r"C:\Users\Olek Pisera\Downloads\8TW8HW_fastq\8TW8HW_reads_over_8kb.fastq"
MIN_READ_LENGTH = 5000
MIN_READS_FOR_CONSENSUS = 4
# ================= PRIMERS (UNIQUE ONLY) =================
# We removed the common linkers.
# Cutadapt will now only look for these specific unique tags.
ROW_PRIMERS = {
"A": "GTTGTGACGTATCA",
"B": "GTTGACAGTGCACA",
"C": "GTTGGTACTGACCA",
"D": "GTTGCGATCAGTCA",
"E": "GTTGATGCACGACA",
"F": "GTTGTGCAGACTCA",
"G": "GTTGGACTACGTCA",
"H": "GTTGCAGTAGACCA"
}
COL_PRIMERS = {
"01": "AGTCAGCTACAGGA",
"02": "AGTCTCGATCACGA",
"03": "AGTCCTGCATAGGA",
"04": "AGTCGATCGTCAGA",
"05": "AGTCACGTACTGGA",
"06": "AGTCTGACTGCAGA",
"07": "AGTCCAGTCGATGA",
"08": "AGTCGTACAGTCGA",
"09": "AGTCATCGACGTGA",
"10": "AGTCGCATGACTGA",
"11": "AGTCTACTAGCGGA",
"12": "AGTCCGTGATACGA"
}
# ================= HELPER FUNCTIONS =================
def win_to_wsl_path(win_path):
if ":" in win_path:
drive, tail = win_path.split(":", 1)
clean_tail = tail.replace('\\', '/')
return f"/mnt/{drive.lower()}{clean_tail}"
return win_path
def rev_comp(seq):
complement = {'A': 'T', 'C': 'G', 'G': 'C', 'T': 'A', 'N': 'N'}
return "".join(complement.get(base, base) for base in reversed(seq.upper()))
def filter_reads(input_fastq, output_fastq, min_length):
total, kept = 0, 0
try:
with open(input_fastq, 'r') as fin, open(output_fastq, 'w') as fout:
while True:
header = fin.readline()
if not header: break
seq = fin.readline()
plus = fin.readline()
qual = fin.readline()
total += 1
if len(seq.strip()) >= min_length:
fout.write(header + seq + plus + qual)
kept += 1
except: return 0, 0
return total, kept
def run_command(cmd):
try:
subprocess.run(cmd, shell=True, check=True, stdout=subprocess.DEVNULL, stderr=subprocess.PIPE)
return True
except subprocess.CalledProcessError:
return False
def get_longest_read_record(fastq_file):
longest_seq = ""
longest_header = ""
max_len = 0
with open(fastq_file, 'r') as f:
while True:
header = f.readline().strip()
if not header: break
seq = f.readline().strip()
plus = f.readline()
qual = f.readline()
if len(seq) > max_len:
max_len = len(seq)
longest_seq = seq
longest_header = header
return longest_header, longest_seq
def generate_consensus(well_name, clean_fastq, work_dir):
header, backbone_seq = get_longest_read_record(clean_fastq)
if not backbone_seq: return None, "No Reads"
backbone_file = work_dir / f"{well_name}_backbone.fasta"
with open(backbone_file, "w") as f:
f.write(f">{well_name}_backbone\n{backbone_seq}\n")
paf_file = work_dir / f"{well_name}.paf"
map_cmd = f"minimap2 -x map-ont {backbone_file} {clean_fastq} > {paf_file}"
try:
subprocess.run(map_cmd, shell=True, check=True, stderr=subprocess.DEVNULL)
except: return None, "Minimap2 Failed"
consensus_file = work_dir / f"{well_name}_consensus.fasta"
racon_cmd = f"racon -t 4 {clean_fastq} {paf_file} {backbone_file} > {consensus_file}"
try:
subprocess.run(racon_cmd, shell=True, check=True, stderr=subprocess.DEVNULL)
except: return None, "Racon Failed"
final_seq = ""
with open(consensus_file, "r") as f:
for line in f:
if not line.startswith(">"): final_seq += line.strip()
if not final_seq: return None, "Racon Empty"
return f">{well_name}_consensus_RACON\n{final_seq}\n", f"Polished ({len(final_seq)} bp)"
# ================= MAIN LOGIC =================
def main():
original_input = Path(win_to_wsl_path(INPUT_FILE_WIN))
work_dir = Path.home() / "consensus_temp_work"
if work_dir.exists(): shutil.rmtree(work_dir)
work_dir.mkdir(parents=True, exist_ok=True)
input_fastq = work_dir / "input.fastq"
demux_dir = work_dir / "demux"
demux_dir.mkdir()
print(f"1. Copying input...")
shutil.copy(original_input, input_fastq)
# 2. GENERATE BARCODES (Unique Only)
barcode_fasta = demux_dir / "barcodes.fasta"
with open(barcode_fasta, "w") as f:
for r_name, r_seq in ROW_PRIMERS.items():
for c_name, c_seq in COL_PRIMERS.items():
well_name = f"{r_name}{c_name}"
# Using linked adapters with NO common sequences.
# Reads must contain these exact unique codes.
adapter_seq = f"{r_seq}...{rev_comp(c_seq)}"
f.write(f">{well_name}\n{adapter_seq}\n")
# 3. RUN CUTADAPT
# Tuned for short, unique barcodes (~14bp)
print("2. Running Cutadapt (Unique Barcodes Only)...")
cutadapt_cmd = (
f"cutadapt "
f"-g file:{barcode_fasta} "
f"-o {demux_dir}/{{name}}_raw.fastq "
f"--revcomp "
# -O 12: Requires 12bp overlap (almost the full unique barcode)
# -e 0.0: Allows ~0 errors in 14bp (reasonable for Nanopore)
# --no-indels: KEPT as requested
f"-e 0.0 --no-indels --discard-untrimmed -O 12 "
f"{input_fastq}"
)
if not run_command(cutadapt_cmd):
print("Cutadapt failed.")
sys.exit(1)
# 4. CONSENSUS LOO
fastq_files = sorted(list(demux_dir.glob("*_raw.fastq")))
print(f"3. Generating Consensus...")
print("\n" + "="*70)
print(f"{'Well':<6} | {'Raw':<6} | {'Valid':<6} | {'Status':<30}")
print("-" * 70)
success_count = 0
final_seqs = {}
for raw_fastq in fastq_files:
well_name = raw_fastq.name.replace("_raw.fastq", "")
clean_fastq = demux_dir / f"{well_name}.fastq"
total_reads, valid_reads = filter_reads(raw_fastq, clean_fastq, MIN_READ_LENGTH)
if valid_reads == 0:
if total_reads > 0:
print(f"{well_name:<6} | {total_reads:<6} | {valid_reads:<6} | {'Skipped (No Valid Reads)':<30}")
continue
if valid_reads < MIN_READS_FOR_CONSENSUS:
print(f"{well_name:<6} | {total_reads:<6} | {valid_reads:<6} | {'Skipped (Low Reads)':<30}")
continue
seq_data, status = generate_consensus(well_name, clean_fastq, demux_dir)
if seq_data:
final_seqs[well_name] = (seq_data, valid_reads)
success_count += 1
print(f"{well_name:<6} | {total_reads:<6} | {valid_reads:<6} | {status:<30}")
print("="*70)
final_output_dir = original_input.parent / "Plate_Consensus_UniqueBarcodes"
if final_output_dir.exists(): shutil.rmtree(final_output_dir)
final_output_dir.mkdir(parents=True, exist_ok=True)
for well, (seq_data, count) in final_seqs.items():
out_file = final_output_dir / f"{well}_consensus_{count}reads.fasta"
with open(out_file, "w") as f:
f.write(seq_data)
shutil.rmtree(work_dir)
print(f"Done! {success_count} sequences saved to 'Plate_Consensus_UniqueBarcodes'.")
if __name__ == "__main__":
main()
I’ve now run this a couple times alongside a traditional single plasmid ($15) confirmation from the same well. It finds the SNPs, and works.
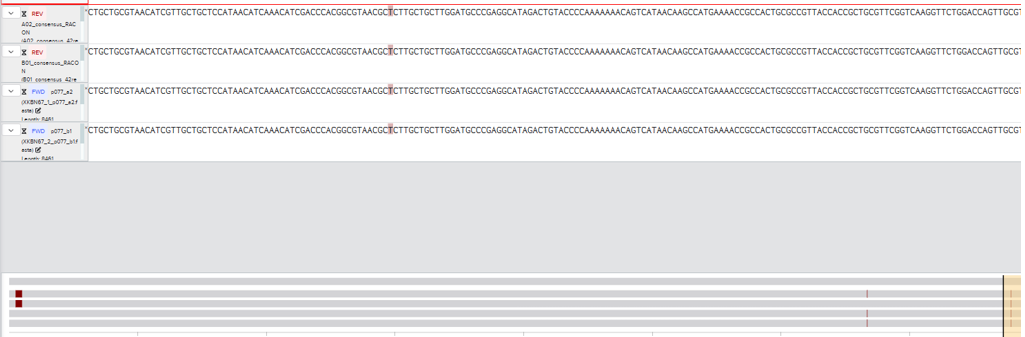
I’m still building trust with this way of doing things, as the 1bp error version of this script burned me with bad alignments, but I’ve now confirmed 3x runs with individual plasmid follow up.
For me, this will save about ~$5-10k per year in sequencing costs, but mostly it will ensure that I never have to decide how badly I need a plasmid the next day. I’ve gotten up to 24x good consensus sequences from 1,000 full length reads, but it can probably be pushed further.